Lab 6. Molecular Phylogenetics
March 12 & 13, 2024
06_biol_200_lab_6.RmdNotes:
Bring your computer to lab with Geneious Prime already installed. Before coming to lab, download and install the trial version software for Geneious Prime from https://manage.geneious.com/free-trial. After sharing your email and other info, click to download the appropriate Geneious Prime Installer for your computer’s operating system. Open the install file after download and complete the installation. Be sure that you keep the information about your subscription, you will need to know the License Key to complete installation.
Complete the Pre-Lab exercise for BEFORE this week’s lab. It can be found HERE
Complete this handout during your lab session AND begin the post-lab exercise BEFORE leaving your lab session this week!
The post-lab exercise is due at the start of Lab 7.
Objectives:
Understand how DNA sequence data can be converted into phylogenetic characters.
Evaluate the similarities and differences between DNA sequence data and morphological data as they are used for phylogenetic inference.
Learn how to query GenBank to find and download DNA sequences.
Learn how phylogenies can be used to understand character evolution.
KEY WORDS: bioinformatics; Sanger sequencing; chromatogram; GenBank; multiple sequence alignment;homology; positional homology; synapomorphy
Overview
Over the past weeks you have learned about characters and character states using real and imagined organisms, and you have learned how to read and construct simple phylogenetic trees. A major goals of today’s lab is to demonstrate how we can use phylogenies to ask and answer questions about the evolution of organisms. Specifically, you will see the power of trees to inform us about character evolution. But first, you should be comfortable with DNA sequence data as a source of phylogenetic characters. Then we will revisit the greenhouse character matrix you created, which we will view in light of both the UPGMA tree as well as a tree based on DNA sequences.
1 Pre-Lab Exercise for Lab 6
Let’s look at the DNA sequence data for our greenhouse taxa from Lab 2 and explore how it can be used to reconstruct the phylogeny of the greenhouse plants. This part of the lab will also introduce you to bioinformatics, or the use of computational methods and tools such as databases for understanding biological data. Bioinformatics is an increasingly important part of all biology.
1A DNA sequence data
Here, a technique called Sanger sequencing was used to label individual nucleotide bases with fluorescent molecules. In Sanger sequencing, each of the four different nucleotides is labeled with a different fluorescent molecule, so that each type of nucleotide base (A, C, G, or T) fluoresces a different color when struck with a laser. A detector records the flash of color emitted as the DNA moves through a polyacrylamide gel one base at a time, and a computer reads the colors and converts the information into a text string of bases; i.e. a sequence of nucleotides (hence, DNA sequence). Most of these steps are performed by a special instrument called an automated DNA sequencer, which needs billions of copies of the DNA sequence of interest, so that the flash of color is bright enough for the detector to see it. We create those billions of copies using PCR (polymerase chain reaction)!
Follow the instructions below to launch Geneious Prime and examine sequences from the greenhouse plants:
-
Download the file “biol200_DNA.geneious” using the button below.
Be sure to save the file to your Desktop . This contains one file of raw Sanger sequence data, as well as some sequences for the taxa we examined a few weeks ago. -
Launch the program Geneious Prime. Once launched, you will see a large window like this:
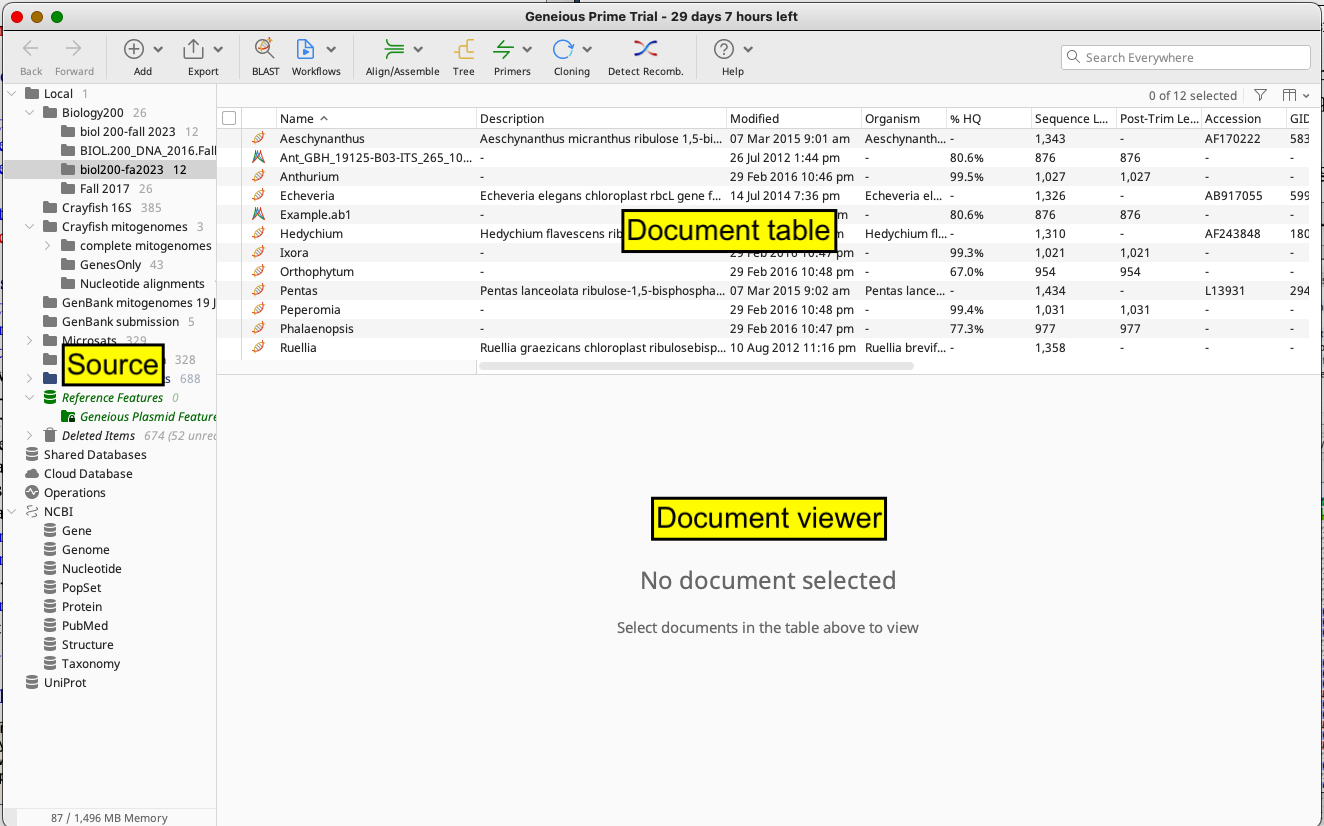
Figure 1: Genieous Prime window on MacOS computers
There’s a lot going on here, so let’s take a second to get oriented to the various windows. We have the Sources Panel, Document Table, and Document Viewer open. Note: you can resize these panels at any time by clicking and dragging the borders between windows.
- ) The Sources Panel contains a series of folders organizing the documents that can be stored and manipulated in Geneious Prime, including any sequences you’re examining. If you click on a folder, its files will appear in the Document Table window.
- ) The Document Table will display documents within a given folder. These are typically DNA sequences but may also be sequence alignments, trees, or other documents that we will not go over today.
- ) Highlighting a file in the Document Table will cause the contents of that file (e.g. the raw DNA sequence data, the DNA alignment, etc.) to be displayed in the Document Viewer. If the Document Viewer is too small, you can resize this window by clicking on the gray bar between the Document Table and Document Viewer (moving the cursor over the bar, a double-headed arrow will appear—move the bar up or down). Double-clicking on a file will open it in a new window.
- ) Within the Document Viewer panel, once you select a document, you should see a Side Panel with a column of tabs, a house icon at the top. This Side Panel Control bar allows you to change the way things appear in the Document Viewer. For example, you can change the colors used for highlighting or stop showing the annotations (bars labeling sequences). Clicking on the icons in that column will expand or minimize the menus associated with each.
- Now we must import our sequence data into Geneious. From the top menu bar, go to Add \(\rightarrow\) Import Files. A popup window will ask you for the file format; choose “Auto Detect Format” and click OK. Navigate to the Desktop and select the “biol200_DNA.geneious” file you downloaded earlier. Then click Import. A number of files should appear in the Document Table, including one file called “Example.ab1”. This is a raw sequence file. If you cannot see enough of the file name, slide the border between the “Name” and “Description” columns to the right until you can see the “Example.ab1”.
-
Now click on the file name and take a look at that raw sequence file. The Document Viewer should look something like this:
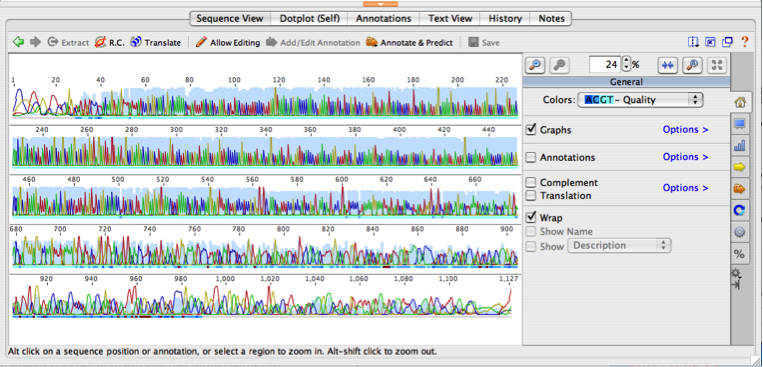
Figure 2: Example of of what raw sequencing data looks like in Geneious
The entire 876 base pair (bp) sequence will appear in the window, so zoom in to see it better. To zoom, click on the button at the right of the sequence with arrows pointing toward each other. The colored peaks represent the raw sequence data generated by the DNA sequencer. All of the peaks together are referred to as a DNA chromatogram. The chromatogram depicts the fluorescence measured by the detector as the laser beam struck the fluorescent-labeled DNA. The automated DNA sequencer interpreted the fluorescent peaks from the chromatogram to generate DNA base calls (nucleotide letters below each peak) and the light blue bar depicts confidence in each base call (higher bar indicates higher confidence that the computer correctly identified that base).
Now, use the scroll bar to the right of the chromatogram to scroll up and down through the whole sequence. You’ll notice that the peaks at the beginning (first 30-40 base pairs) and the end (last 200-300 bp) of the sequence are broad and messy looking (and consequently the blue confidence bars are low; sharp, narrow peaks = high quality base calls). This pattern is completely normal and results from a technical limitation of Sanger sequencing.
In contrast to the raw sequence file, the files containing sequence data for the greenhouse taxa have been downloaded from GenBank. GenBank is a worldwide repository for all DNA sequence data that is generated during the course of scientific studies, and is a free, open resource maintained by the National Center for Biotechnology Information (NCBI). Researchers who generate DNA sequence data are required to deposit their sequences here when publishing scientific articles. GenBank has become an enormous resource containing trillions of base pairs of DNA sequence data.
Specifically, the sequences for the greenhouse taxa are for the gene rbcL, which is a chloroplast gene that codes for the large subunit of the ribulose bisphosphate carboxylase/oxygenase enzyme, usually called RuBiSCO. This protein incorporates carbon dioxide into a 5-carbon sugar molecule during the Calvin Cycle of photosynthesis (incredibly important for life on earth).
Examining your rbcL sequences, you will note that, unlike the raw sequence data, they only have the sequence of bases (no chromatogram) and have been annotated (labeled) to show what region they represent. The annotations can be especially helpful when looking at a sequence that spans both exons and introns by displaying where different regions in the DNA begin and end. Take a moment to look through some of the other files in the Document Table, which will give you a chance to get comfortable with using Geneious Prime.
1B Aligning DNA sequences
Now that we’ve taken a look at our DNA sequence data, let’s revisit how to convert DNA data to phylogenetic trees. When we did UPGMA using the character state matrix, we used characters to aid in reconstructing phylogeny—i.e., we relied on similarities and differences in character states among taxa to generate an estimate of phylogeny.
Although we didn’t discuss it explicitly at the time, a critical
prerequisite to scoring morphological character states for phylogenetic
reconstruction is that we have to be sure to compare
Question 1. With this in mind, consider comparing characters of bird wing structure and insect wing structure for phylogenetic purposes. Would this comparison be problematic? Why or why not?
Just like with morphological characters, only homologous DNA characters can be compared. Let’s think about our rbcL data. All plants have an rbcL gene, because the common ancestor of all plants possessed this gene. So by comparing rbcL sequences among plants, we are evaluating homologous DNA sequences. But how do we define characters and character states within our rbcL sequences? For DNA, the position of the nucleotide within a gene is always the character (a concept called positional homology). For example, in our rbcL sequences, we would want to compare the nucleotide present at the 100th position in this gene for all taxa. Consequently, in DNA, the position is the character, and the four possible nucleotides (A, T, C, G) are the character states.
Question 2. If a gene has 1500 base pairs, how many characters are present in the gene?
To reinforce the concepts above, let’s look at a simple character state matrix for some of our greenhouse taxa, with examples of both morphological and rbcL DNA characters that you scored. The point of showing the matrix below is to emphasize that there is no fundamental difference in how we treat DNA sequence characters compared to any other character.
| Table 1: Character table for greenhouse plant species including both morphological and DNA characters | ||||||
| Taxon | Morphological Characters | DNA Characters | ||||
|---|---|---|---|---|---|---|
| Leaf Venation | Number of Petals | Flower Symmetry | Alignment Position 142 | Alignment Position 169 | Alignment Position 437 | |
| Anthurium | net-veined | 0 | radial | C | T | T |
| Begonia | net-veined | 4-5 | bilateral | G | C | G |
| Euphorbia | net-veined | 0 | radial | T | T | A |
| Ixora | net-veined | 4 | radial | A | C | A |
| Phalaenopsis | parallel | 3 | bilateral | G | T | T |
| Tillandsia | parallel | 3 | radial | C | T | T |
When we assemble our rbcL data into a character state matrix for analysis, it is crucial to line up the sequences so as to maintain positional homology—a process called multiple sequence alignment. Let’s perform such an alignment using our rbcL data.
In Geneious, select all of the rbcL files by holding down the apple key (on a Mac) or control key (on a PC) and clicking on them. All of the sequences should appear in the Document Viewer. Zoom in on the sequences until you can read the individual bases. Notice that the sequences don’t all begin (or end) with the same exact stretch of bases, although if you look carefully you can find the same pattern of bases in all the sequences. Just like you saw at the extreme ends of your chromatogram, factors related to PCR and Sanger sequencing can affect the quality and lengths of sequences across taxa, especially at the extreme ends.
With all of the rbcL sequences selected, click on the “Align/Assemble” button in the top toolbar. In the menu that appears, click the ‘Multiple Align’ option. When a new popup window appears, make sure that the “Geneious Alignment” button is selected (no need to change anything else); then click OK. After a few seconds, a new “Nucleotide alignment” file will be created in the Document Table; it should also appear automatically in the Document Viewer.
Take a moment to scroll through the alignment. All of the different
sequences should now be perfectly lined up;
i.e.
Question 3. What is the total length of the alignment? Hint: To find this, look in the Document Table. \(\space\) ______________
When opening an alignment file, by default Geneious highlights the bases in the alignment that differ from other bases at the same position. This setting should already be on, and most of the bases in the alignment will look gray (i.e. unhighlighted) because all taxa share the same character state for those positions. However, if most of your bases are still very colorful, change the highlighting disagreements setting:
- Turn on Highlighting Disagreements: In the Document Viewer, open the tab with the house icon on the Side Panel. In the window that appears, find the “Highlighting” button and make sure it is selected. Then click on the blue “Options” arrow to the right. This will call up additional buttons and drop-down menus. Make sure the two drop-down menus under “Highlighting” are set to “Disagreements” and “Consensus”.
Once the proper Highlighting is turned on, scroll through the sequence alignment again and note the differences among taxa. More similar sequences should be grouped together in the alignment list. Do you see any differences that are shared among taxa?
Look at character 460. Which taxa share character state C? \(\space\) \(\space\) ____________________________
Look at character 1,051. Which taxa share character state G? \(\space\) \(\space\) ____________________________
2 Comparing DNA and Morphological Data
A “total evidence” phylogeny based on DNA sequence data from many loci (including rbcL) has already been constructed for you— obtain the Greenhouse Morphology and Tree Sheet. Additionally, this sheet contains a morphology-based UPGMA tree for all 10 greenhouse taxa and the morphological data matrix from Lab 2. If you are interested in learning about the many ways to build a phylogeny, consider taking Prof. Moore’s Plant Systematics course (BIOL 323/324), offered in the spring.
Find Anthurium and Peperomia in the UPGMA morphological tree, the morphological data matrix (on the Green-house Morphology and Tree Sheet), and the rbcL alignment, to answer the following questions:
Anthurium and Peperomia are sister in the morphological tree, indicating that they share similar character states. Do they share great similarity in DNA sequence too? Look through the alignment and count the number of sequence differences between these two taxa for only the first 500 bases in the alignment (don’t count the first 18 where no sequence is available for Anthurium). To facilitate determining these differences, make these two taxa adjacent to one another in the alignment by clicking on the desired taxon name in the Document Viewer and dragging it up or down.
- Total number of differences = _____________ out of the first 500 characters
-
Consider whether Anthurium and Peperomia share any unique sequence changes in the rbcL alignment. Search through the alignment for any changes that are
shared between these two taxa but areNOT present in any other taxa; if we saw many of these kinds of unique shared bases, it would be evidence of their close relationship. An example of a character state that is unique to Anthurium and Peperomia is given in the table below. Can you find any more? Explain why you might have expected this result, based on the position of Anthurium and Peperomia in the total evidence tree.Uniquely shared by Anthurium and Peperomia: Character number 583 Character state C - Based on the above DNA results, it would appear that Anthurium and Peperomia are not very closely related. Instead, what clade does the total evidence tree suggest that Anthurium is more closely related to?
In the UPGMA tree, Aloe, Tillandsia and Phalaenopsis are distantly related but in the total evidence DNA tree, they form a clade. This suggests that despite being morphologically very different, they might have a lot in common in DNA. Let’s see how many shared differences we can find.
-
Go to the rbcL alignment and fill in the table below with characters (nucleotide positions) in which Aloe, Tillandsia, and Phalaenopsis share a unique character state with respect to all other taxa in the alignment. Look at the example first:
Uniquely shared by Tillandsia, Aloe, and Phalaenopsis: Character Number Character State Character Number Character State - With your group, discuss which data—morphology or DNA—you find more convincing regarding the relationships of Anthurium. Which line of evidence do you think is more likely to be correct? Think about the advantages and disadvantages of using each type of data. Explain your reasoning below.
- If the phylogeny based on DNA is correct, how do you explain the presence of flowers lacking petals in both Anthurium and Peperomia? Propose a hypothesis to explain this.
You may have noticed, in the above questions, that we placed a strong emphasis on character states uniquely shared by a group of taxa. The goal in these questions is to find derived character states shared among taxa (remember that shared, derived characters are known as synapomorphies). These types of character states can inform phylogenetic relationships, whereas ancestral character states cannot provide evidence of phylogenetic relationships.
- Within tetrapods (four-legged vertebrates), is the presence of hair a shared, derived character state? Explain your answer.
We can learn much about character evolution given a phylogeny that we trust, based on extensive morphological and molecular data, by “mapping” the morphological character states onto our total evidence tree. To map character states, we indicate the relevant character state for a given character next to the taxon name in a tree. The entire greenhouse morphology matrix has been mapped onto the total evidence tree on the phylogeny handout; i.e., the entire first row of this matrix indicates the character states for Anthurium, the second row is for Cattleya, and so on. Use this sheet to answer the following questions.
-
Based on morphological mapping onto the total evidence tree (ignore the UPGMA tree), let’s determine how many times the following character states evolved. The number of evolutionary origins will be easy to determine for some of these character states but it will be harder to determine for others. Do your best and don’t hesitate to ask for guidance if you’re not sure!
Parallel Leaf Venation ___________ Presence of Leaf Trichomes ___________ Opposite Leaves ___________ Absence of Flower Pedicel ___________ 5-petalled Flowers ___________ 3-petalled Flowers ___________ 0-petalled Flowers ___________ Bilateral Symmetry ___________ - Now consider which of the six morphological characters seem to be relatively poor predictors of phylogeny. In other words, which of the characters seem to be prone to multiple origins (or losses) of the same character states?Write the character name(s) [not the character state names] below.
-
Based on the relationships depicted in the total evidence tree, what do you think the most likely ancestral character state is for all of the taxa in the tree, for the following characters?
Leaf Venation ___________ Leaf Trichomes ___________ Leaf Arrangement ___________ Floral Symmetry ___________ - Discuss with your group: what do you think the ancestral character state for number of petals is, for all taxa in the tree? Try to figure this out. Why is it so difficult to determine this?
- So, if we were interested in determining the ancestral number of petals in all of angiosperms (not just the 10 taxa here), what would you suggest that we do, from a phylogenetic point of view? Below, suggest a brief plan to resolve this question.
-
Explore the relative values of morphological vs. DNA data for
reconstructing phylogeny, with these questions:
-
Only some of the morphological characters you scored were predictive of
phylogeny, resulting in discrepancies between the morphological and
total evidence trees. How could you create an improved
morphological data matrix? - Do you think morphology or DNA might be more prone to subjectivity in scoring character states? Explain your answer briefly.
- When dealing with morphological data, we must ensure that we compare homologous structures, and with DNA, we must ensure positional homology. Do you think it is easier to establish homology in DNA or morphology, and why?
- Which do you think provides a greater number of potential characters—morphology or DNA?
-
Only some of the morphological characters you scored were predictive of
phylogeny, resulting in discrepancies between the morphological and
total evidence trees. How could you create an improved
- Create a UPGMA tree for the rbcL alignment in Geneious. Click on your alignment in the Document Table and then click on the “Tree” icon. A popup menu will appear; leave the default settings except for the “Tree Build” method—set this to UPGMA, then click on OK. Compare this tree to the total evidence tree. How is it different?
2A Greenhouse Questions
When your instructor tells you it’s OK to go, be sure to take this handout with you up to the greenhouse, along with the Greenhouse Morphology and Tree Sheet.
Take a minute or two to locate the plants on the total evidence phylogeny. All the plants in this tree (except Anthurium, Peperomia, and Euphorbia) fall into two major groupings: one with 4 or 5 petals per flower and the other with 3 or 6 petals per flower. The former group is part of a large clade of angiosperms known informally as the dicots; most dicots have flower parts (sepals, petals, stamens, etc.) that occur in multiples of 4 or 5. The 3- and-6-petalled plants are part of another clade known informally as monocots; in contrast to dicots, monocots have flower parts that occur in multiples of 3. Anthurium belongs to the monocots while Peperomia and Euphorbia belong to the dicots. While Anthurium superficially resembles Euphorbia/Peperomia (both have reduced flowers that lack petals), this is due to convergent evolution.
The monocots and dicots are among the largest clades of flowering plants—collectively, they include ∼95% of all 300,000 angiosperm species (and with the exception of the ferns and a few other plants, nearly all of the plants in the greenhouse as well). Below are several questions about monocots and dicots in light of our morphological and phylogenetic data, as well as a couple of other questions about the plants and characters you’ve used—discuss and answer these questions with your group.
-
As mentioned above, all the monocots have flower parts in 3’s. What other character from your morphology matrix diagnoses all monocots except Anthurium?
___________________________________ -
Using what you now know about monocots and dicots, find one more of each in the greenhouse, and make sure to note in which room it is found.
Monocot: _________________________________ Dicot: _________________________________
Go to the last room of the greenhouse and look at the desert-adapted plants in this room. Do you notice how many of them are succulent (i.e. they have fleshy water-storage organs)? For example, there are cacti (which are dicots) and agaves (which are monocots), along with many other plants in many other distantly related monocots and dicots.
- These morphological features are independently derived in these different groups through convergent evolution. During their separate evolutionary histories, distantly related taxa developed similar adaptations to arid envi-ronments. How would this contribute to the placement of these taxa on a phylogenetic tree constructed from a morphological character matrix? Explain briefly below.
- Thinking in ecological terms, why might it be good to avoid characters like succulence when searching for characters to use in a morphological phylogenetic analysis? Explain your answer below.
- Alternatively, what might it indicate about the evolutionary history of a group if you found such environmentally specific character states in a taxon that was not living in that particular kind of environment?
-
- Which morphological characters that you scored in Lab 2 do you think might be prone to convergence?
3 Coda: The Importance of Tree-Thinking
After completing these exercises, we hope that you have become adept at reading and interpreting phylogenetic trees, and that you have a much clearer appreciation for the importance of tree thinking within biology. As you have seen, robust estimates of phylogeny using large data sets are critical for many fields of biology, because they provide the necessary comparative framework for understanding the origin and evolution of characters, from morphological and molecular characters to ecology and biogeography. Phylogenies also frequently cause us to propose new hypotheses about character evolution when unexpected relationships are discovered, as you saw when trying to understand convergent evolution. Without strongly supported phylogenetic inference, much of the story of life on Earth would be difficult to tell.
4 Post-Lab Assignment for Lab 6 (due at start of Lab 7)
Where are the genes? Visualizing organellar genomes in Geneious Prime
To get oriented to gene and genome data within Geneious Prime, let’s look at the lettuce chloroplast genome. Click on the file “lettuce chloroplast genome” in the Document Table. A circular genome map with colored arrows will appear in the Document View (bottom window). The arrows represent annotations. Geneious displays different classes of annotations with different colors, and the arrows indicate the 5’$$3’ direction of the annotation. Green arrows denote the locations of genes, and yellow arrows indicate the locations of coding sequences (“CDS”), which only apply to protein-coding genes. Hot pink arrows are tRNAs, and red arrows indicate rRNAs. Take a moment to look at the different kinds of genes.
- Do all parts of the genome code for proteins or RNAs? Can these non-coding regions have functions? If yes, what kind of functions might these non-coding regions have?
For some protein-coding genes, there are multiple CDS’s for the same gene. This indicates that the gene is composed of more than one exon, with introns between the CDS’s. You may notice that thin yellow lines connect each exon, this is the way Geneious depicts exons that are part of the same gene.
- Find the clpP gene—how many introns are present? _______________
- How many exons? _______________
You can also see the amino acid translation for CDS’s by clicking on the Translation button on the Side Panel of the Document View. Scroll around and look at different kinds of protein-coding genes. Different amino acids are different colors, and the one-letter standard abbreviation for the amino acid is provided. Find the start and stop codons for several genes. Stop codons are black with an asterisk.
- In genes with introns, why is there no stop codon at the end of the first exon?
- Find the rbcL gene and write down the amino acid sequence
for the first six amino acids (use the one-letter abbreviation):
______________________________
What about mitochondrial genomes? Use the “modern human mitochondrial genome” included in your dataset to answer the following questions:
- Which has a longer total length—the lettuce chloroplast genome or
the human mitochondrial genome? How much longer?
___________________________________________________________________________
- What types of genes are present in BOTH genomes? To answer this,
look for the following specific classes of genes: protein-coding genes,
rRNA genes, and tRNA genes. Write your answer below:
__________________________________________________________________________________________
- Are there any introns in the human mitochondrial genome? ______________________________
Variation among genomes
Now let’s look at genomic variation, both within and among species, starting with human mitochondrial genome diversity. Animal mitochondrial genomes evolve rapidly, which is useful for evolutionary analyses and forensics! Find the files for the Homo sapiens mitochondrion. There are 4 sequences, one from Africa, one from Europe, one from Australia/New Guinea, and one from Oceania. Look in the description to identify them.
To compare these four genomes, you need to align them so that the homologous genes are in the same positions. Because the mitochondrial genomes are much larger than the rbcL sequences, the alignment will take longer and so we’d like to use a different method than Geneious for alignment. Instead, let’s install a Plugin that will allow for faster processing. Go to Tools \(\rightarrow\) Plugins. In the popup box, scroll to find MAFFT and install it. Then close the window. Select all four human mitochondrial genomes, go to the “Align/Assemble” button, and click on “Pairwise/Multiple Align…”. In the box that pops up, make sure that “MAFFT Alignment” is selected at the top of this menu, and click OK. After a bit (this will take longer than the rbcL alignment), a new document will appear that will show all four genomes aligned to one another.
On the right-hand side of the Document View check the box called “Highlighting” to see the differences among aligned sequences; all positions that are identical are grayed out. When this box is checked, zoom in on the alignment. (Also, make sure that annotations are turned on.)
- What kinds of patterns do you see in the differences among the genomes? Are they concentrated in any particular regions? Are there any insertions/deletions?
- If the differences are in protein-coding gene regions, do they result in amino acid sequences for that gene? (Hint: turn on Translation if it’s not already turned on.)
- Which region of the genome has the most differences? Does this region have any genes in it?
As the most variable part of the mitochondrial genome, this region is very important in systematics, population genetics, human forensics, and genealogy. This rich mitochondrial variation can be used not only with human populations, but other hominids, primates, and even more distantly related animals! To explore these genomic differences, we have mitochondrial genomes for two extinct taxa of the genus Homo—Neanderthals and Denisovans. These taxa inhabited the colder parts of Eurasia, and both went extinct in the past 50,000 years. They were so closely related to Homo sapiens that they interbred with populations of H. sapiens, as evidenced in our own nuclear genomes today. Unless your ancestry is 100% sub-Saharan African, between 1-4% of your genome (on average) is composed of sequences that were inherited from Neanderthals and Denisovans. We now have complete genomes from multiple individuals of both Neanderthals and Denisovans isolated from bone fragments found in Europe and central Asia. The techniques for recovering and sequencing DNA from such bones, are extremely challenging and this achievement is one of the triumphs of science over the past decades.
So, how different were Neanderthals and Denisovans from modern humans? Select the mitochondrial genomes for these two taxa and the four modern humans, then align all six genomes in the same manner as before.
What do you see? How much variation is there in modern humans compared to the two extinct taxa? To make this easier to visualize, set one of the modern human genomes as a reference sequence and change the highlighting settings to show differences to the reference: choose any of your modern human genomes, then right click (or control-click) on the sequence name in the Document View (not the Document Table). This will call a popup menu; click “Set as reference sequence”, then save the document (“Save” button at the top of the Document View). Make sure the “Highlighting” settings are set to show differences from the Reference Sequence.
Now, find the mitochondrial genome for a chimpanzee (Pan troglodytes). Make an alignment between any human genomes you want and the chimpanzee genome. How different are they?
- Let’s look for different kinds of mutations in your mitochondrial alignments. Using the tools at your disposal (for example, turning on Translation), look for examples of the following kinds of mutations and fill in the table below:
| Kind of mutation | Position in alignment | If this is in a protein-coding gene, does this cause a change in amino acid sequence? Is it a missense, nonsense, or frameshift? |
| Substitution | ||
| Indel |